
这项研究发现,HIV 并不是被动地“躲进”休眠的 T 细胞,而是在感染后的 72 小时内主动重编程宿主细胞,启动 KLF2 和 p53 通路、压制增殖因子 MYC,让细胞进入静止状态,从而把自己和细胞一同隐藏起来,形成难以清除的潜伏库。
这一机制颠覆了长期的认知,也为未来通过干预这条通路来阻断或清除 HIV 潜伏提供了新的思路。

核心发现:HIV感染激发T细胞进入“休眠”状态
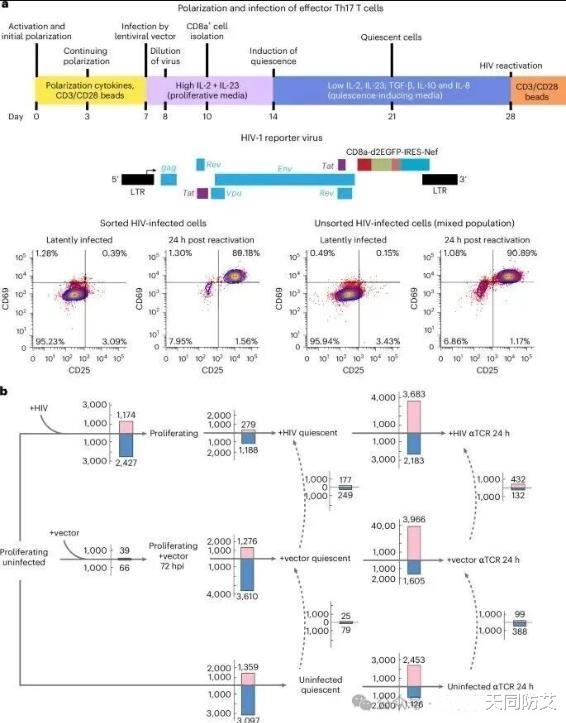
最近的研究揭示了一个不为人所知的机制:HIV病毒并非简单地在干细胞中建立潜伏,而是主动地、主动地“劫持”并重编程CD4+ T细胞,使其进入静止或“休眠”(静止)状态,从而为病毒自身创造一个长期的、受保护的潜伏库(潜在储库)。
暂停状态对病毒的生存至关重要,因为在状态下,抗逆转录病毒治疗(ART)无法有效清除这种病毒。
病毒如何重编程T细胞?
研究发现,HIV感染会引发一系列转录组的变化,这意味着细胞的内基因表达模式的全面改变。这些变化激活了两个关键的细胞信号采集:
p53因子:p53因子是一种著名的肿瘤抑制基因,在细胞早期和DNA损伤时被激活,并能诱导细胞进入生长信号或信号通路。研究表明,HIV会激活p53因子,这有助于病毒实现其目标。
KLF2基因:这是一个诱导细胞进入静止状态的关键调节因子。研究发现,HIV感染会显著上调KLF2的表达水平。
更重要的是,这种上调不是激活的,而是依赖于HIV病毒整合到另一个基因组中。当研究人员使用一种名为拉替拉韦(raltegravir)的药物阻止病毒整合时,KLF2的水平就不会上升。
这表明不可实现的良好作用是病毒实现两个潜伏的关键。研究发现,同时激活p53和KLF2全部的细胞,其内部HIV病毒的转录活性最低,这表明病毒已经成功进入了深度潜伏状态。
关键“牺牲品”:MYC基因
为了让细胞进入休眠状态,HIV必须关闭细胞的增殖开关。研究发现,HIV感染导致了MYC基因及其相关信号减弱的显著降低。MYC是一个驱动细胞增殖和增殖的关键基因。当MYC被萎缩后,细胞的增殖、抑制和翻译活动就会减慢,导致细胞最终进入静止状态。
为了验证这一发现,研究人员进行了“MYC基因敲除”实验,结果发现,单独的MYC基因就足以模拟HIV感染引起的细胞急剧组变化和增殖减慢。这说明MYC的休眠是HIV感染诱导T细胞休眠的一个核心环节。
对HIV感染者的意义
最近的研究提供了关于艾滋病毒病毒如何建立和维持其潜伏库的新观点。以下是几个关键的启示:
潜伏库的“先天性”形成:病毒在感染CD4+ T细胞后,并非偶然地进入潜伏,而是通过一套设计的分子机制激发促使细胞进入休眠,从而保证自身的持续存在。这解释了为什么即使接受了有效的ART治疗,潜伏库仍然难以被根除。
新的靶点:既然病毒通过激活p53和KLF2并纠缠MYC来实现潜伏,那么针对这些分子的新疗法可能会为清除潜伏库提供新的策略。例如,开发出抑制KLF2或p53的药物,以阻止病毒进入休眠状态。相反,如果能重新激活MYC,或许可以“唤醒”潜伏的病毒,在ART的攻击中诱发暴露。
Tat蛋白的潜在作用:研究还指出,HIV的Tat蛋白可能在诱导这种休眠程序中发挥关键作用,因为它能够独立于其他病毒蛋白诱导类似的效果。这为未来的研究指明了方向,即研究Tat蛋白如何具体地调节细胞信号信号通路。
总的来说,这项研究极大地加深了我们对HIV潜伏库的认识,从被动的“躲藏”到主动机制的“重编程”。它为我们战胜HIV潜伏库、实现功能性治愈提供了新的希望和思路。
文章标题:HIV infection reprogrammes CD4+ T cells for quiescence and entry into proviral latency
作者:Leah M. Plasek Hegde, Lalith S. Gunawardane,et al.
DOI:10.1038/s41564-025-02128-y
编译:松鼠哥