·安全性:必须符合相关安全标准,包括电气安全、生物相容性、辐射安全等。
·有效性:必须具备经科学验证的预期治疗效果。
·质量管理体系:制造商必须建立完善的质量管理体系,确保产品质量和安全性。
·标识和说明书:必须标明生产厂商、产品名称、型号、规格、生产日期等信息,并配备详细的使用说明书。
二、不同种类医疗器械的适用法规
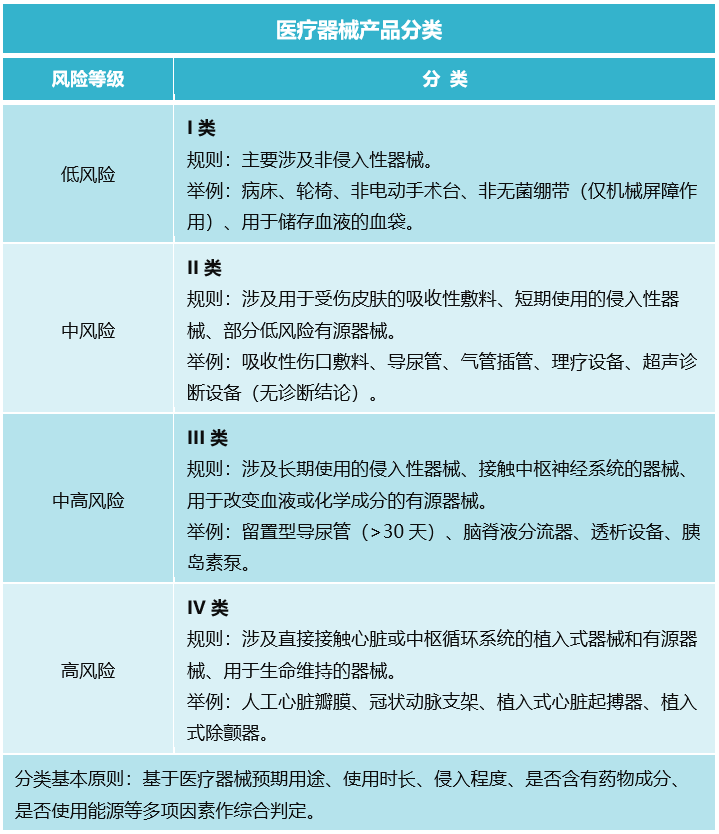

四、不同分类医疗器械对应的注册路径
·I类和II类器械 - Cadastro(登记)路径
相对简化的流程,主要由第三方认证机构(Organismo Certificador de Produtos - OCP)负责审核与批准。
流程分解为:
-准备技术文档
-提交至OCP
-OCP技术分析
-OCP批准与发证
-上市后监督
特点: 流程更快、成本较低,审核主体是OCP而非ANVISA本身。
·III类和IV类器械 - Registro(注册)路径
更严格的审批流程,全部申请必须由ANVISA总部直接审核与批准。
流程分解为:
-准备技术文档
-提交至ANVISA
-ANVISA初步分析
-技术分析与临床评估
-良好生产规范GMP核查
-ANVISA批准与发证
-上市后监督
特点:流程复杂、耗时漫长(可能长达数年)、成本较高,且高度依赖强有力临床证据。

·A类和B类IVD - 通知路径 (Via de Notificação)
类似于医疗器械的Cadastro,但流程更规范。
·A类(低风险):制造商提交符合性声明,声明其产品符合巴西法规要求。通过系统通知ANVISA后即可上市,ANVISA进行事后监督。
·B类(中低风险):
需提交技术文档至OCP或ANVISA(根据法规细则),审核通过后获得授权。
·C类和D类IVD - 注册路径 (Via de Registro)
较严格的、由ANVISA主导的审批流程。
流程分解为:
-性能评估:核心过程。技术文档必须包含↘
·分析性能研究:证明检测方法的精确度、准确度、灵敏度、特异性、检测限等;
·临床性能研究:证明在目标人群中的临床灵敏度和特异性,通常需提供基于巴西人群和本地流行菌株/病毒的临床性能数据。
·稳定性研究:包括效期稳定性、开瓶稳定性。
-提交与审核:申请提交至ANVISA,ANVISA重点审核性能评估数据的充分和科学性。
-批准与上市后性能跟踪:获批后制造商必须执行上市后性能跟踪,持续收集和评估产品在真实世界中的性能数据,并定期向ANVISA报告。