在单细胞分析过程中有两个极其重要的环节,一个是前期的质控,一个便是降维聚类后的细胞类型注释。
生物学家使用术语细胞类型来表示在数据集中稳健的细胞表型,根据特定标记物(即蛋白质或基因转录物)的表达进行识别,并且通常与特定功能相关。例如,血浆 B 细胞是一种白细胞,可分泌用于对抗病原体的抗体,可以使用特定标记物进行识别。了解样品中的细胞类型对于了解数据至关重要。例如,了解肿瘤中存在特定的免疫细胞类型或骨髓样本中存在不寻常的造血干细胞,可以深入了解您可能正在研究的疾病。
然而,与任何分类一样,类别的大小和它们之间的边界部分是主观的,并且会随着时间的推移而变化,例如,因为新技术允许更高分辨率的细胞视图,或者因为被认为没有生物学意义的特定“亚表型”被发现具有重要的生物学意义。因此,细胞类型通常进一步分为“亚型”或“细胞状态”(例如活化与静息),一些研究人员使用术语“细胞身份”来避免有时对细胞类型、细胞亚型和细胞状态的武断区分。
同样,多种细胞类型可以是单个连续体的一部分,其中一种细胞类型可能会转变或分化为另一种细胞类型。例如,在造血中,细胞从干细胞分化为特定的免疫细胞类型。尽管经常在这种分化的早期和晚期之间划出硬边界,但这些细胞的状态可以更准确地通过分化程度较低和分化程度较高的细胞表型之间的分化坐标来描述。
代码复现
首先过滤掉一些不影响代码的弃用和性能警告:
import warningswarnings.filterwarnings("ignore", category=DeprecationWarning)import numbafrom numba.core.errors import NumbaDeprecationWarning, NumbaPendingDeprecationWarningwarnings.simplefilter("ignore", category=NumbaDeprecationWarning)
加载包:
import scanpy as scimport pandas as pdimport numpy as npimport osfrom scipy.sparse import csr_matriximport seaborn as snsimport matplotlib.pyplot as pltimport celltypistfrom celltypist import modelsimport scarches as scaimport urllib.request
再次过滤pandas的不必要警告:
warnings.filterwarnings("ignore", category=pd.errors.PerformanceWarning)
设置图窗参数:
sc.set_figure_params(figsize=(5, 5))
随后我们导入之前的umap降维聚类后的数据并用于后续分析执行细胞类型注释的经典方法是基于已知与特定细胞类型相关的单个或一小部分标记基因。这种方法可以追溯到 “pre-scRNA-seq times”,当时单细胞数据是低维的(例如,基因面板由不超过 30-40 个基因组成的 FACS 数据)。这是一种快速透明的数据注释方式。然而,当特定细胞类型不存在唯一标记时(通常是这种情况),这种方法可能会变得更加复杂,甚至更不客观,因为正确注释需要标记或表达阈值的组合。一组强大的标记基因和先验知识或注释经验可以在这方面提供帮助,但这种方法存在决策不明确和主观的风险。
在此设置中,数据通常在 annotation 之前进行聚类,因此我们可以对 cells 组进行 Comments,而不是进行每个 cell 的调用。这不仅更省力,而且对噪声的抵抗力更强:单个单元格可能没有特定标记的计数,即使它是在该单元格中表达的,这仅仅是由于单个单元格数据固有的稀疏性。聚类能够检测整体基因表达高度相似的细胞,因此可以解释单细胞水平的缺失。
最后,有两个角度可以接近基于标记基因的注释。一种选择是从数据中预期的所有细胞类型的标记基因表中工作,并检查这些簇的表达。另一种选择是检查哪些基因在您定义的簇中高表达,然后检查它们是否与已知的细胞类型或状态相关。当然也可以在这些方法之间来回切换。
在收集最开始的标记物表的时候,有几个需要注意的点:
●以前研究特定细胞类型和亚型并报告这些细胞类型的标记基因的论文。但其中蛋白质水平的标记物(例如用于 FACS)有时在转录组数据中效果不佳,因此使用基于 RNA 的论文中的标记物通常更有可能起作用。
●有时一个数据集中的标记在其他数据集中效果不佳。因此,理想情况下,在多个数据集中验证标记集。
●最后,与专家合作通常是有用的:作为生物信息学家,尝试与对组织、生物学、预期细胞类型和标记等有更广泛了解的生物学家合作
接下来我们先定义一个marker集:
marker_genes = {"Proximal tubular cells": ["Slc22a6", "Slc22a8", "Slc12a3", "Aqp1", "Cdh16", "Clcn5", "Gstm1", "Ppargc1a", "Lrp2", "Scl22a18"], # 近端小管细胞"Distal tubular cells": ["Slc12a1", "Nkcc2", "Aqp2", "Slc4a1", "Kcnj1", "Atp6v1b1", "Kcnj10", "Tmem27", "Clcnka", "Slc26a3"], # 远端小管细胞"Podocytes": ["Nphs1", "Nphs2", "Wt1", "Podxl", "Cd2ap", "Synpo", "Actn4", "Nephrin", "Slit2", "Fyn"], # 足细胞"Macrophages": ["F4/80", "Cd64", "Cd206", "Mrc1", "Tmem119", "Cx3cr1", "Ccr2", "Il1b", "Nos2", "Tnf"], # 巨噬细胞"Dendritic cells": ["Clec9a", "Cst3", "Ly75", "Batf3", "Flt3", "Xcr1", "Ccr7", "Cd11c", "Cd80", "Cd86"], # 树突状细胞"Natural Killer Cells": ["Cd56", "Nkg7", "Ifng", "Gzma", "Fcer1g", "Klrg1", "Tyrobp", "Il15", "Tbet", "Perforin"], # 自然杀伤细胞"Neutrophils": ["Mpo", "Ly6g", "Cd11b", "Cxcr2", "Il17a", "Tlr4", "Ccl3", "Ccl4", "Mmp8", "Rhoh"], # 中性粒细胞"T Cells (CD4+)": ["Cd4", "Cd3e", "Il7r", "Ccr7", "Cd25", "Tcrb", "Tcra", "Foxp3", "Cd28", "Il2ra"], # CD4+ T细胞"T Cells (CD8+)": ["Cd8a", "Cd8b", "Gzmk", "Gzma", "Cd3e", "Tcrb", "Tcra", "Ifng", "Cd28", "Fasl"], # CD8+ T细胞"B Cells": ["Cd19", "Ms4a1", "Ighm", "Cd20", "Igk", "Iglc1", "Prdm1", "Bcl6", "Pax5", "Cd22"], # B细胞}
我们将遍历所有细胞类型,只保留在 adata 对象中找到的基因作为该细胞类型的标记。这将防止我们开始绘图时出现错误:
marker_genes_in_data = dict()for ct, markers in marker_genes.items(): markers_found = list() for marker in markers: if marker in adata.var.index: markers_found.append(marker) marker_genes_in_data[ct] = markers_found
我们还是选择scran标准化数据用于分析:
adata.X = adata.layers["scran_normalization"]
设置特征基因:
adata.var["highly_variable"] = adata.var["highly_deviant"]
继续PCA分析:
sc.tl.pca(adata, n_comps=50, use_highly_variable=True)
计算邻域图:
sc.pp.neighbors(adata)
umap:
sc.tl.umap(adata)
我们对巨噬细胞单独观察:
macrophage_cts = [ "Macrophages",]
for ct in macrophage_cts: print(f"{ct.upper()}:") # print cell subtype name sc.pl.umap( adata, color=marker_genes_in_data[ct], vmin=0, vmax="p99", # set vmax to the 99th percentile of the gene count instead of the maximum, to prevent outliers from making expression in other cells invisible. Note that this can cause problems for extremely lowly expressed genes. sort_order=False, # do not plot highest expression on top, to not get a biased view of the mean expression among cells frameon=False, cmap="Reds", # or choose another color map e.g. from here: https://matplotlib.org/stable/tutorials/colors/colormaps.html ) print("\n\n\n") # print white space for legibility

Macrophage
即使是单个细胞类型的标记物也经常在不同的数据子集中表达,即单个标记物通常不在单个细胞类型中唯一表达。相反,正是这些子集的交集将告诉您感兴趣的细胞类型在哪里。
您可能会注意到的另一件事是,标志物通常表达稀疏,即通常只是检测到标志物的细胞类型的细胞子集。这是由于 scRNA-seq 数据的性质:我们只对细胞中 RNA 分子总量的一小部分进行测序,并且由于这种二次采样,我们有时不会对细胞中特定基因的转录本进行采样,即使它们在该细胞中表达。因此,我们不根据例如一组标记的最小表达阈值来注释单个细胞。相反,我们首先通过聚类将数据细分为相似细胞组(即“分区”数据),从而解释单个基因的“缺失转录本”,而不是根据整体转录组相似性进行分组。然后,我们可以根据它们的整体标记表达模式来注释这些簇。
接下来用气泡图展示一下主要在哪些群表达:
macrophage_markers = { ct: [m for m in ct_markers if m in adata.var.index] for ct, ct_markers in marker_genes.items() if ct in macrophage_cts}
sc.pl.dotplot( adata, groupby="leiden_res0_25", var_names=macrophage_markers, standard_scale="var", # standard scale: normalize each gene to range from 0 to 1)

气泡图
可以看到主要在5,11两个群表达,我们看一下其分布:
sc.pl.umap(adata, color="leiden_res0_25",legend_loc="on data")

umap图
如果要针对巨噬细胞进行后续分析,很明显可以单独挑出5,11两个群分析。
接下来我们注释到图中:
cl_annotation = {"5": "Macrophage","11": "Macrophage",}
adata.obs["manual_celltype_annotation"] = adata.obs.leiden_res0_25.map(cl_annotation)
sc.pl.umap(adata, color=["manual_celltype_annotation"])
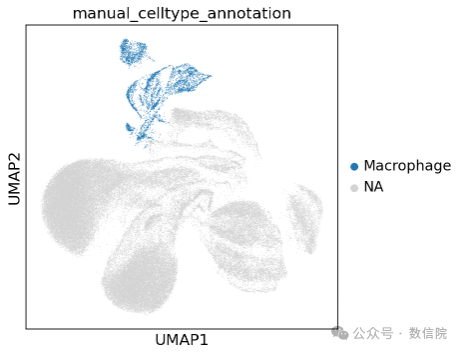
注释结果图