转自:医学界
重症肌无力(MG)是一种乙酰胆碱受体(AChR)抗体介导为主的神经肌肉接头(NMJ)自身免疫性疾病,约85%的全身性疾病患者和50%~60%的眼部疾病患者AChR抗体阳性[1]。目前认为,抗AChR抗体的总抗体滴度与临床严重程度无绝对相关性,且存在显著个体差异,对此,近期四川省人民医院谈颂教授团队发表了一篇题为《乙酰胆碱受体抗体在重症肌无力中的研究进展》综述,从AChR结构、抗体致病机制、检测局限等多维度展开分析,为MG的精准诊疗提供了全新视角[1]。
AChR:既是神经肌肉传递的“关键开关”,也是抗体攻击的“靶心”
要理解AChR抗体的致病逻辑,首先需明确AChR的核心功能。作为NMJ后膜上的“信号接收器”,成熟型AChR是由2个α1、1个β1、1个δ、1个ε亚基组成的五聚体配体门控离子通道,其表面的2个乙酰胆碱(ACh)结合位点是神经信号传递的“关键开关”:当神经末梢释放的ACh结合这些位点后,AChR会打开中央阳离子通道,引发钠离子内流,最终触发肌肉收缩。
值得注意的是,由于不同患者的抗体可识别不同亚基和不同表位,AChR的“免疫原性”和MG的抗体反应呈现高度异质性。针对不同AChR亚基和表位的抗体反应构成了表位水平的免疫异质性,直接影响抗体的致病潜力与致病机制:
传统观点认为,α1亚基的“主要免疫原区(MIR)”是抗体攻击的主要靶点,因此临床检测抗MIR抗体可能有助于评估症状严重程度并辅助区分MG亚型。
最新高分辨率结构研究还发现,MG患者的抗体还可靶向β1、δ等多个亚基及α1亚基的非MIR区域。其中α亚基特异性抗体的存在与病情活动度密切相关,而β亚基特异性抗体可能相对“良性”,即使量高也不易引起严重症状。
除单个表位外,有研究通过体外重组抗体组合实验发现,靶向不同AChR亚基的抗体联合可显著增强补体激活效果,最强协同效应出现在α亚基IgG1抗体与非α亚基抗体联合时。
因此,AChR抗体的致病性不仅取决于滴度,更与其表位特异性及协同机制相关。未来或许可将患者抗AChR抗体的表位分布、内化动力学、补体激活效率等整合,建立反映抗体整体致病能力的“致病指数”,以指导个体化治疗策略和精准预测病情进展。
三大致病机制协同“作乱”,单一机制难解释临床异质性
AChR抗体并非通过“单一模式”致病,而是通过补体激活、受体阻断、抗原内化三大机制协同作用,从不同层面破坏神经冲动传递。
■补体激活:最主要的“破坏路径”
补体介导的突触后膜损伤是AChR抗体最重要的致病机制之一。当补体激活型抗体结合AChR后,其Fc段会招募补体C1q,启动经典补体级联反应,最终形成膜攻击复合物(MAC)。MAC插入肌细胞膜后造成局部膜结构破坏和AChR丢失,引起AChR密度下降、神经肌肉接点突触间隙扩大、终板电位幅度降低等病理改变,导致神经冲动传递失败,引起肌无力表现。
一项研究在2年内纵向收集了50名AChR抗体阳性全身性MG患者血清样本(n=210),以检测补体激活、受体阻断、抗体内化的频率。结果显示,补体激活是最普遍的机制,可在84.8%的样本中被观察到,其次是抗原内化(63%)和受体阻断(30.4%),补体激活与抗原内化同时存在的比例为45.6%,补体激活与受体阻断作用同时存在的比例为10.8%,三种病理机制同时活跃的比例为17.4%。仅出现受体阻断作用的比例为2.1%。(图1)。该研究还发现,抗体与AChR的结合能力、抗体介导的病理机制(补体激活、抗原内化和受体阻断作用)随时间波动,并且因患者而异,同时纵向变化表明,部分患者临床症状的严重程度与AChR自身抗体变化/病理机制的波动性呈现相关性[2]。
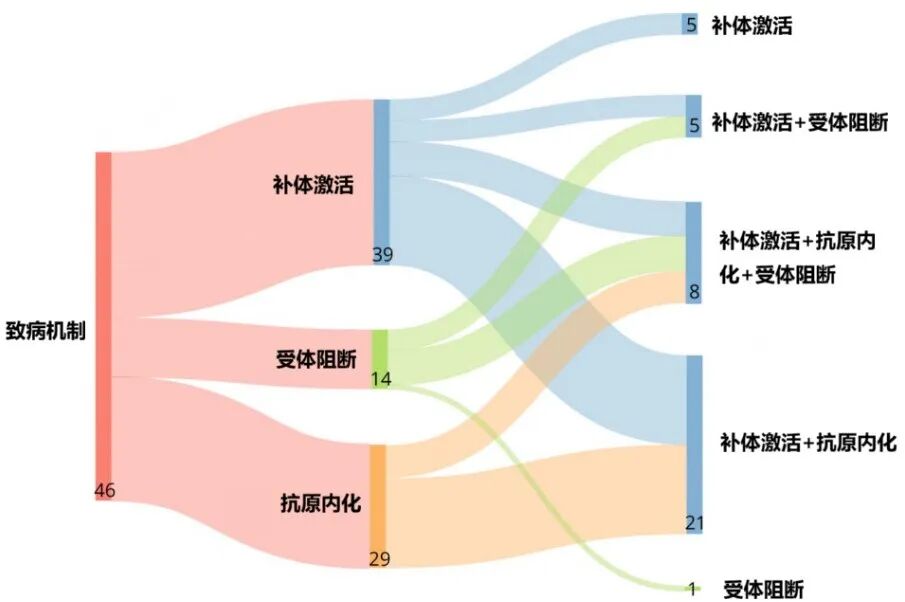
AChR抗体阳性全身性MG中不同致病机制比例
有研究显示,在检测到AChR自身抗体的样本中,59.7%可检测到MAC形成,而MAC沉积伴随AChR密度下降,进一步支持其致病性[3,4]。此外C5a的水平还可能与疾病严重程度相关[5]。这也解释了为何靶向C5的抑制剂可通过阻止C5裂解成C5a和C5b片段,从而阻止MAC形成,从而快速改善MG患者症状,降低疾病复发风险[6]。
■受体阻断:直接“掐断”信号传递
部分AChR抗体会直接结合AChR的功能域,通过两种方式阻断信号传递:一是竞争结合ACh的结合位点,从而阻断ACh与受体的结合;二是物理堵塞离子通道,阻止阳离子内流。一项研究中这类“阻断型抗体”在30.4%的患者血清中被检出[2],同时存在阻断型与补体激活型抗体的患者,更易发展为全身型MG,疾病严重程度较高,预后较差,且常伴有胸腺瘤。值得注意的是,AChR具有2个Ach结合位点,仅当抗体结合到这些关键结构或其附近产生空间阻碍时,才可真正发挥阻断作用。这也解释了阻断效应的强度并不完全取决于抗体浓度,而更多与表位位置及亲和力相关。
■抗原内化:悄悄“清除”受体
AChR抗体还能通过“交联-内吞”机制减少膜表面AChR数量:IgG1/IgG3等双价抗体会同时结合两个相邻的AChR,将其“交联”成簇,触发肌细胞的内吞作用,最终导致AChR被降解。63%的AChR-MG患者存在这一机制,且45.6%的患者同时合并补体激活,形成“双重打击”。值得关注的是,IgM和IgA也能介导AChR内化,因此各类抗体都可以参加抗原调节。
多种致病机制的共存在AChR抗体阳性的MG患者中十分常见,因此未来临床治疗应结合患者的机制分型,如通过功能性抗体分析(补体活性检测、阻断试验、内化评估等),考虑不同作用机制的药物联合治疗,提高疗效并减少无效治疗负担。
■抗体的“身份标签”很重要:Ig亚类与表位决定致病能力
抗AChR抗体的致病性不仅取决于“作用机制”,还与其“身份标签”,即抗体Ig亚类和表位特异性密切相关。在Ig亚类中:
IgG抗体是MG中最主要的致病抗体类型。其中IgG1和IgG3是AChR-MG中的“主力致病者”,在67.4%和21.7%的全身型MG患者中可检出。二者均具备强大的补体激活能力,可通过经典途径诱导MAC形成。
IgM抗体虽占比仅12%,但作为一种五聚体结构,单个IgM分子就携带10个结合位,其启动补体的效率远超IgG,是经典途径中最有效的触发者之一,同时IgM可高效诱导AChR交联和内化,效率为IgG的2.3倍。因此,即使IgM型抗体在患者体内含量不高,仍可加重局部补体攻击和受体丢失。
IgA型抗体占比10%,其不依赖新生儿Fc受体(FcRn)介导的再循环,故不受血浆置换或FcRn抗剂治疗影响,这可能解释了部分患者对常规治疗反应欠佳的现象。
综上,抗AChR抗体的Ig类别和亚类决定了其致病机制偏向。IgG1/IgG3介导者致病性最强,IgM通过高效补体活化加剧病情,而IgA则常规治疗耐受性较强。因此,未来可在初诊阶段进行抗AChR抗体的亚类分型,有助于精细制定个体化治疗方案。
结语
AChR抗体的复杂性与异质性,决定了MG诊疗不能再依赖“一刀切”的模式。从识别AChR的结构特征,到解析三大致病机制的协同作用,再到突破传统检测局限,医学研究正逐步实现从“经验性治疗”向“精准化管理”的转变。随着抗体功能检测技术的成熟与个体化治疗策略的完善,MG患者的精准诊疗时代”已不再遥远。
参考文献:
[1]郭雨鹤,谈颂.乙酰胆碱受体抗体在重症肌无力中的研究进展[J].华西医学,2025(6).
[2]Khani-HabibabadiF,RoyB,PhamMC,etal.AChRAutoantibodyPathogenicPropertiesAreHeterogeneouslyDistributedandUndergoTemporalChangesAmongPatientsWithMyastheniaGravis.NeurolNeuroimmunolNeuroinflamm.2025Sep;12(5):e200436.
[3]ObaidAH,ZografouC,VadysirisackDD,etal.Heterogeneityofacetylcholinereceptorautoantibody-mediatedcomplementactivityinpatientswithmyastheniagravis.NeurolNeuroimmunolNeuroinflamm,2022,9(4):e1169.
[4]HuangYF,SandholmK,PerssonB,etal.Visualizationandcharacterizationofcomplementactivationinacetylcholinereceptorantibodyseropositivemyastheniagravis.MuscleNerve,2024,70(4):851-861.
[5]AguirreF,ManinA,FernandezVC,etal.C3,C5aandantiacetylcholinereceptorantibodyasseveritybiomarkersinmyastheniagravis.TherAdvNeurolDisord,2020,13:1756286420935697.
[6]HowardJFJr,UtsugisawaK,BenatarM,etal.Safetyandefficacyofeculizumabinanti-acetylcholinereceptorantibody-positiverefractorygeneralisedmyastheniagravis(REGAIN):aphase3,randomised,double-blind,placebo-controlled,multicentrestudy.LancetNeurol,2017,16(12):976-986.
本材料由阿斯利康提供,仅供医疗卫生专业人士进行医学科学交流,不用于推广目的。
审批编号:CN-173524过期日期:2026-3-2